Gestalt diagnosis of children with dysmorphism - Necessity for establishing genetic diagnostic approach
Abstract
Background: The overall prevalence of intellectual disability is approximately 2-3% in the general population and can be caused by genetic and environmental factors. Genetic factors include chromosomal anomalies, single-gene disorder, deregulation of imprinted genes, and multiple malformation syndromes without an identified genetic basis and idiopathic. In Bangladesh, the genetic diagnosis of dysmorphic patients has not yet been well established. Therefore, gestalt diagnosis has a crucial role in establishing the differential diagnosis, management, counseling, and genetic diagnostic approach.
Aim of the study: The aim of the present study was to assess the effectiveness and necessity of gestalt diagnosis on the suspected genetic syndrome with intellectual disability and comorbidities.
Methods: This prospective study was conducted at Dhaka Shishu Hospital during the period from December 2017 to May 2018. The study included 21 children with intellectual and developmental disabilities (IDD) who attended OPD and Mental Health Clinic of Dhaka Shishu Hospital, Dhaka, Bangladesh. Elaborate history taking, physical examination, and psychological assessment were done. The dysmorphic features were analyzed and correlated with syndromic diagnosis using OMIM search. Parents were counseled about the preferred genetic diagnostic tests to confirm the syndromic diagnosis. Prognosis of the index children and chance of recurrence in the next pregnancy was discussed when a particular syndrome was suspected. Informed written consent was taken from parents of every patient to use the photograph and data for diagnostic and academic purposes.
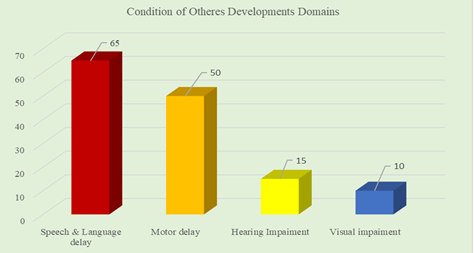
Results: Among total participants, 40% had severe cognitive delay 35% had a moderate delay, 25% had a mild cognitive delay, 70% had behavior problems and 55% had ASD and/or ADHD features. The seizure was present in 35% of patients. Among other comorbidities; speech and language delay was in 65%, motor delay was in 50%, vision impairment was in 10%, hearing impairment was 15 %. Suspected cases were, Noonan syndrome: 4, Angelman syndrome: 3, Fragile X syndrome: 2, Kabuki syndrome: 2, Sotos syndrome: 2. Genetic diagnosis could be established in only 2 patients with suspected fragile X syndrome.
Conclusion: The study emphasizes the necessity to approach gestalt diagnosis in syndromic children with IDD along with locally available low-cost genetic diagnostic facility thereby increasing the possibility of providing appropriate management and/or genetic counseling.
Downloads
References
Allanson JE, O’Hara Farkas LG, Nair RC: Anthropometric craniofacial pattern profiles in Down syndrome. Am J Med Genet 1993, 47:748-752.
Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A: Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum Mutat 2013, 34:1589-1596.
Gravholt, Claus H., et al. "Turner syndrome: mechanisms and management." Nature Reviews Endocrinology 15.10 (2019): 601-614.
Allanson JE, Hall JG, Hughes HE, Preus M, Witt RD: Noonan syndrome: the changing phenotype. Am J Med Genet 1985, 21:507-514.
Bokinni Y: Kabuki syndrome revisited. J Hum Genet 2012, 57:223-227.
Dixon MJ: Treacher Collins syndrome. Hum Mol Genet 1996, 5:1391-1396.
Read, Andrew P., and Valerie E. Newton. "Waardenburg syndrome." Journal of medical genetics 34.8 (1997): 656-665.
Williams CA: Neurological aspects of the Angelman syndrome. Brain Dev 2005, 27:88-94.
Fridman C, Kok F, Koiffmann CP: Hypotonic infants and the Prader-Will Syndrome. J Pediatr (Rio J) 2000, 76:246-250.
Mollison L, O’Daniel JM, Henderson GE, Berg JS, Skinner D. Parents’ perceptions of personal utility of exome sequencing results. Genet. Med. (2020) 22:752-7. doi: 10.1038/s41436-019-0730-8
Lingen M, Albers L, Borchers M, Haass S, Gärtner J, Schröder S, et al. Obtaining a genetic diagnosis in a child with disability: impact on parental quality of life. Clin Genet. (2016) 89:258-66. doi: 10.1111/cge. 12629
Reiff M, Bernhardt BA, Mulchandani S, Soucier D, Cornell D, Pyeritz RE, Spinner NB. “What does it mean”: uncertainties in understanding results of chromosomal microarray testing. Genet Med. (2012) 14:250-8. doi: 10.1038/gim.2011.52
Krabbenborg L, Vissers LELM, Schieving J, Kleefstra T, Kamsteeg EJ, Veltman JA, et al. Understanding the psychosocial effects of WES test results on parents of children with rare diseases. J Genet Couns. (2016) 25:1207-14. doi: 10.1007/s10897-016-9958-5.
Curry CJ, Stevenson RE, Aughton D, et al. Evaluation of mental retardation: recommendations of a consensus conference- American College of Medical Genetics. Am J Med Genet. 1997; 72:468 -477. 1
Shevell MI. The evaluation of the child with a global develop- mental delay. Semin Pediatr Neurol. 1998; 5:21-26.
Cassel EJ. The nature of suffering and the goals of medicine. N Engl J Med. 1982; 306:639 - 645.
Dykens EM, Smith AC: Distinctiveness and correlates of maladaptive behaviour in children and adolescents with Smith-Magenis syndrome. J Intellect Disabil Res 1998, 42:481-489.
Martelli-Junior H, Coletta RD, Miranda RT, Barros LM, Swerts MS, Bonan PR. Orofacial features of Treacher Collins syndrome. Med Oral Patol Oral Cir Bucal. 2009;14: E344-8. [PubMed] [Google Scholar]
Trainor PA, Dixon J, Dixon MJ. Treacher Collins syndrome: Etiology, pathogenesis and prevention. Eur J Hum Genet. 2009; 17:275-83. [PMC free article][PubMed] [Google Scholar]
Netchine, I. et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell–Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J. Clin. Endocrinol. Metab. 92, 3148–3154 (2007).
Wollmann, H. A., Kirchner, T., Enders, H., Preece, M. A. & Ranke, M. B. Growth and symptoms in Silver–Russell syndrome: review on the basis of 386 patients. Eur. J. Pediatr. 154, 958–968 (1995).
Price, S. M., Stanhope, R., Garrett, C., Preece, M. A. & Trembath, R. C. The spectrum of Silver–Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J. Med. Genet. 36, 837–842 (1999).
Wakeling, E. L. Silver–Russell syndrome. Arch. Dis. Child 96, 1156–1161 (2011).
Toutain, A, Silver-Russell syndrome (2007) Orphanet http://www.orpha.net/consor/cgi-bin/Disease
Yakoreva, M. et al. A retrospective analysis of the prevalence of imprinting disorders in Estonia. Eur. J. Hum. Genet. 23 (Suppl. 1), 325 (2015).
Noonan JA. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am. J. Dis. Child. 1968; 116:373-380. [PubMed] [Google Scholar]
Nora JJ, Nora AH, Sinha AK, et al. The Ullrich-Noonan syndrome (Turner phenotype). Am J Dis Child. 1974; 127:48-55. [PubMed] [Google Scholar]
Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet C Semin Med Genet. 2003; 117C:57–65. [PubMed] [Google Scholar]
Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010; 42:790–793. [PMC free article] [PubMed] [Google Scholar]
Sil A,Panigrahi A, Visual Dermatology: Waardenburg Syndrome Type II. Journal of cutaneous medicine and surgery. 2020 May/Jun [PubMed PMID: 32421428]
Grewal PS, Knight H, Michaelides M, Asymmetric choroidal hypopigmentation in a Son and mother with Waardenburg syndrome type I. Ophthalmic genetics. 2020 Jun [PubMed PMID: 32281454]
Ren SM, Kong XD, Wu QH, Jiao ZH, Chen C, Qin ZB, [Analysis of genetic variation in patients with Waardenburg syndrome type Ⅱ by next generation sequencing]. Zhonghua yi xue za zhi. 2020 Mar 24 [PubMed PMID: 32234158]