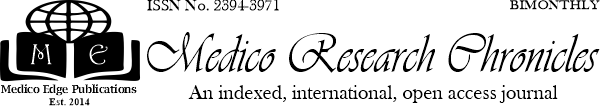
Study of endocrinological complications in children with beta-thalassemia major: A hospital base study.
Abstract
Background: Beta thalassemia major (TM) is the most common inherited genetic disorder worldwide. Patients are at risk of iron overload, which leads to various forms of tissue damage, including endocrinopathies.
Aim of the study: The aim of this study was to evaluate the prevalence and risk factors of endocrine disorders in Adolescent patients.
Methods: A prospective analytical study done was done at Bangladesh Shishu Hospital & Institute, Dhaka, Bangladesh from 1st October 2021 to 1st July 2022 among the diagnosed cases of Beta Thalassemia Major.
Result: Among 80 TM children, 32(44.19%) patients were female and 48(55.81%) were male. The mean (SD) patient age at the time of the study was 11.34 (2.27), range: 2–18 years and 11.12 (2.01) range: 2–12 in males and females, respectively. In those who received combined therapy, 6 of 17 (32.14%) cases had endocrine disorders, compared with 34 of 63 (53.97%) cases who did not have endocrine disorders.
Conclusion: Endocrinological complications in adolescent children with thalassemia major were mild in severity in our study and observed when the serum ferritin went beyond 2500 ng/ml. Early identification and multidisciplinary management are necessary for early recognition,treatment as well as prevention of endocrinological complicationsin patients with Thalassemia Major.
Downloads
References
2. Mentzer WC, Kan YW. Prospects for research in hematologic disorders: sickle cell disease and thalassemia. JAMA. 2001 Feb 7;285(5):640-2.
3. El-Beshlawy A, Kaddah N, Rageb L, Hussein I, Mouktar G, Moustafa A, Elraouf E, Hassaballa N, Gaafar T, El-Sendiony H. Thalassemia prevalence and status in Egypt. Pediatric research. 1999 May;45(5):760-.
4. Viprakasit V, Lee-Lee C, Chong QT, Lin KH, Khuhapinant A. Iron chelation therapy in the management of thalassemia: the Asian perspectives. International journal of hematology. 2009 Nov;90(4):435-45.
5. Porter JB. Monitoring and treatment of iron overload: state of the art and new approaches. InSeminars in hematology 2005 (Vol. 42, No. 2, pp. S14-S18).
6. Gomber S, Dabas A, Bagmar S, Madhu SV. Glucose homeostasis and effect of chelation on β cell function in children with β-thalassemia major. Journal of Pediatric Hematology/Oncology. 2018 Jan 1;40(1):56-9.
7. Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, Saned MS, El-Chafic AH, Fasulo MR, Cappellini MD. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood, The Journal of the American Society of Hematology. 2010 Mar 11;115(10):1886-92.
8. Li CK, Chik KW, Wong GW, Cheng PS, Lee V, Shing MM. Growth and endocrine function following bone marrow transplantation for thalassemia major. Pediatric Hematology and Oncology. 2004 Jan 1;21(5):411-9.
9. Breda L, Gambari R, Rivella S. Gene therapy in thalassemia and hemoglobinopathies. Mediterranean Journal of Hematology and Infectious Diseases. 2009 Nov 16;1(1):e2009008-.
10. Perera NJ, Lau NS, Mathews S, Waite C, Ho PJ, Caterson ID. Overview of endocrinopathies associated with β‐thalassaemia major. Internal medicine journal. 2010 Oct;40(10):689-96.
11. Hejazi S, Safari O, Arjmand R, Qorbani M, Pourrostami K, Safari A, Hemmati A. Effect of combined versus monotherapy with deferoxamine and deferiprone in iron overloaded thalassemia patients: a randomized clinical trial. International Journal of Pediatrics. 2016;4(6):1959-65.
12. Skarmoutsou, C., Papassotiriou, I., Traeger-Synodinos, J., Stamou, H., Ladis, V., Metaxotou-Mavrommati, A., Stamoulakatou, A. and Kanavakis, E., 2003. Erythroid bone marrow activity and red cell hemoglobinization in iron sufficient beta-thalassemia heterozygotes as reflected by soluble transferrin receptor and reticulocyte hemoglobin in content. Correlation with genotypes and Hb A (2) levels. haematologica, 88(6), pp.631-636.
13. Saffari F, Mahyar A, Jalilolgadr S. Endocrine and metabolic disorders in β-thalassemia major patients.
14. Chahkandi T, Norouziasl S, Farzad M, Ghanad F. Endocrine disorders in beta thalassemia major patients. International Journal of Pediatrics. 2017;5(8):5531-8.
15. Najafipour F, Aliasgarzadeh A, Aghamohamadzadeh N, Bahrami A, Mobasri M, Niafar M, Khoshbaten M. A cross-sectional study of metabolic and endocrine complications in beta-thalassemia major. Annals of Saudi medicine. 2008 Sep;28(5):361-6.
16. Farmaki K, Tzoumari I, Pappa C, Chouliaras G, Berdoukas V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. British journal of haematology. 2010 Feb;148(3):466-75.
17. De Sanctis V, Soliman AT, Canatan D, Yassin MA, Daar S, Elsedfy H, Di Maio S, Raiola G, Corrons JL, Kattamis C. Thyroid disorders in homozygous β-thalassemia: current knowledge, emerging issues and open problems. Mediterranean journal of hematology and infectious diseases. 2019;11(1).
18. Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997 Feb 1;89(3):739-61.
19. GS C, MK S. Pattern of Growth Retardation and Sexual Maturation in Children having Beta Thalassaemia. Journal of Nepal Paediatric Society. 2016 Jan 1;36(1).
20. De Sanctis V, Soliman AT, Elsedfy H, Skordis N, Kattamis C, Angastiniotis M, Karimi M, Yassin MA, El Awwa A, Stoeva I, Raiola G. Growth and endocrine disorders in thalassemia: The international network on endocrine complications in thalassemia (I-CET) position statement and guidelines. Indian journal of endocrinology and metabolism. 2013 Jan;17(1):8.